Domanda solo apparentemente stupida. Facendo oggi un giro sui media italiani direi che "chemio" è il termine con cui chi non ha nessuna cognizione di causa definisce in genere i farmaci antitumorali.
Siccome non sono un ragazzino direi che per me la chemio(terapia) è quell'insieme di farmaci che iniziarono ad essere usati per la cura di pazienti oncologici tra anni '60 e '70: gli antiblastici, cioè in genere farmaci che inibiscono la riproduzione cellulare. La classe comprende alchilanti, antimetaboliti, antimitotici e i cosiddetti antibiotici antitumorali il cui capostipite è la doxorubicina. La loro tossicità è tale che richiede misure di sicurezza per il personale sanitario che li deve manipolare. E costituiscono quella classe di composti responsabili di una certa cattiva reputazione dei farmaci oncologici.
Qua sopra si è parlato spesso e a lungo di farmaci oncologici, quindi non vorrei ripetermi. Quindi preferisco analizzare il segno "chemioterapia".
In quest'ottica "chemio" è la croce che devi portare per salvarti o quello che devi fuggire ad ogni costo. E' l'ennesima polarizzazione tra quel moralismo medico che ormai è comunissimo e l'approccio negazionista-complottista. In questo regime simbolico e in questa contrapposizione il concetto di trade-off semplicemente non esiste più. Trade-off: ok, la terapia esiste ma il suo peso e i suoi costi sono tali che preferisco rifiutarla (i costi possono essere la perdita di un arto o di un'altra parte del corpo o quella della funzionalità sessuale o della capacità riproduttiva, per fare alcuni esempi).
Ritornando a "chemio", il problema è che essendo segno (o simulacro) non ha alcun rapporto con la realtà, mentre le patologie sono reali, la qualità della vita è reale, la morte è reale.
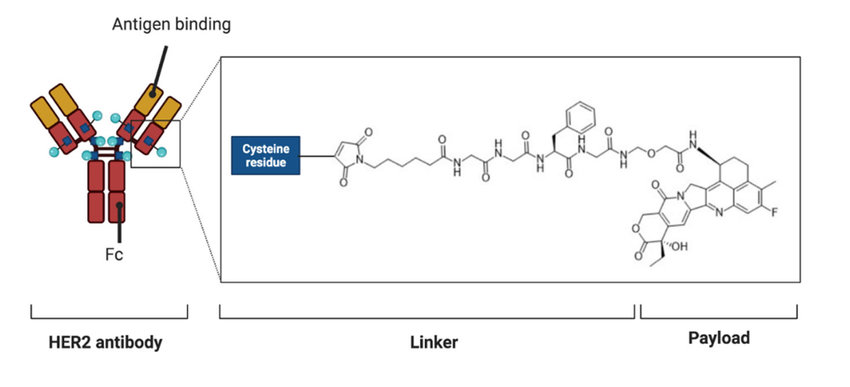
Forse non è inutile ripetere che negli ultimi 25 anni sono statei approvate decine di farmaci targeted (cioè meno tossici perché non colpiscono indistintamente ogni cellula che si sta riproducendo), inibitori di checkpoint, che impediscono alle cellule tumorali di sfuggire al sistema immunitario del soggetto, e alcune terapie cellulari. Non si tratta di terapie o farmaci privi di effetti collaterali, ma siamo molto lontani dalla tossicità degli antiblastici. In alcuni casi siamo rimasti a come erano le cose 40 anni fa, in altri no. E la comunicazione medica al riguardo me la ricordo estremamente carente, specialmente sui social dove, per fare un esempio, il "tumore di Angelina Jolie" era diventato l'immagine standard, con il suo corredo di amputazioni preventive. E in certi casi la comunicazione non è affatto cambiata, anche se oggi abbiamo in Europa ben 4 inibitori PARP (targeted) che hanno cambiato le prospettive per quel tipo di tumore (principalmente per il carcinoma ovarico BRCA+). Ma non solo: vecchi antiblastici di seconda o terza generazione, non mirati, sono diventati targeted, riusati con la tecnologia di coniugazione farmaco-anticorpo (l'anticorpo trasporta il farmaco tossico sulla cellula tumorale, dove viene rilasciato). E EMA ormai ha approvato una decina di ADC (Antibody Drug Conjugate).
Il punto è che, nonostante ogni progresso, ogni episodio che finisce sui media ripropone il simulacro, in cui la chemio-segno equivale alla scienza-segno. Che è un ottimo modo per non parlare di supporto al paziente e, soprattutto, dell'accesso alle cure. Un problema, quello dell'accesso alle cure, che convive senza problemi con un altro oggetto della rappresentazione mediatica, i viaggi della speranza. I viaggi della speranza sono proprio un fenomeno che nasce dal mancato accesso alle terapie, ai farmaci e alle sperimentazioni più avanzate.
Tutto questo accade in un contesto, quello italiano, in cui sicuramente esistono ancora direttori sanitari che ritengono che il cisplatino sia un'ottima cosa, perché costa quanto le patate. Una posizione che tra l'altro interiorizza il principio aberrante che in Italia non possa esistere una sanità pubblica che non sia una sanità dalle risorse sempre più scarse. La spesa sanitaria italiana continua a scendere in rapporto al PIL. Il problema non è l'assenza di risorse pubbliche: è la scelta politica di allocarle verso altri capitoli di spesa - armamenti inclusi - piuttosto che garantire l'accesso universale alle terapie oncologiche innovative.


.png)
.png)